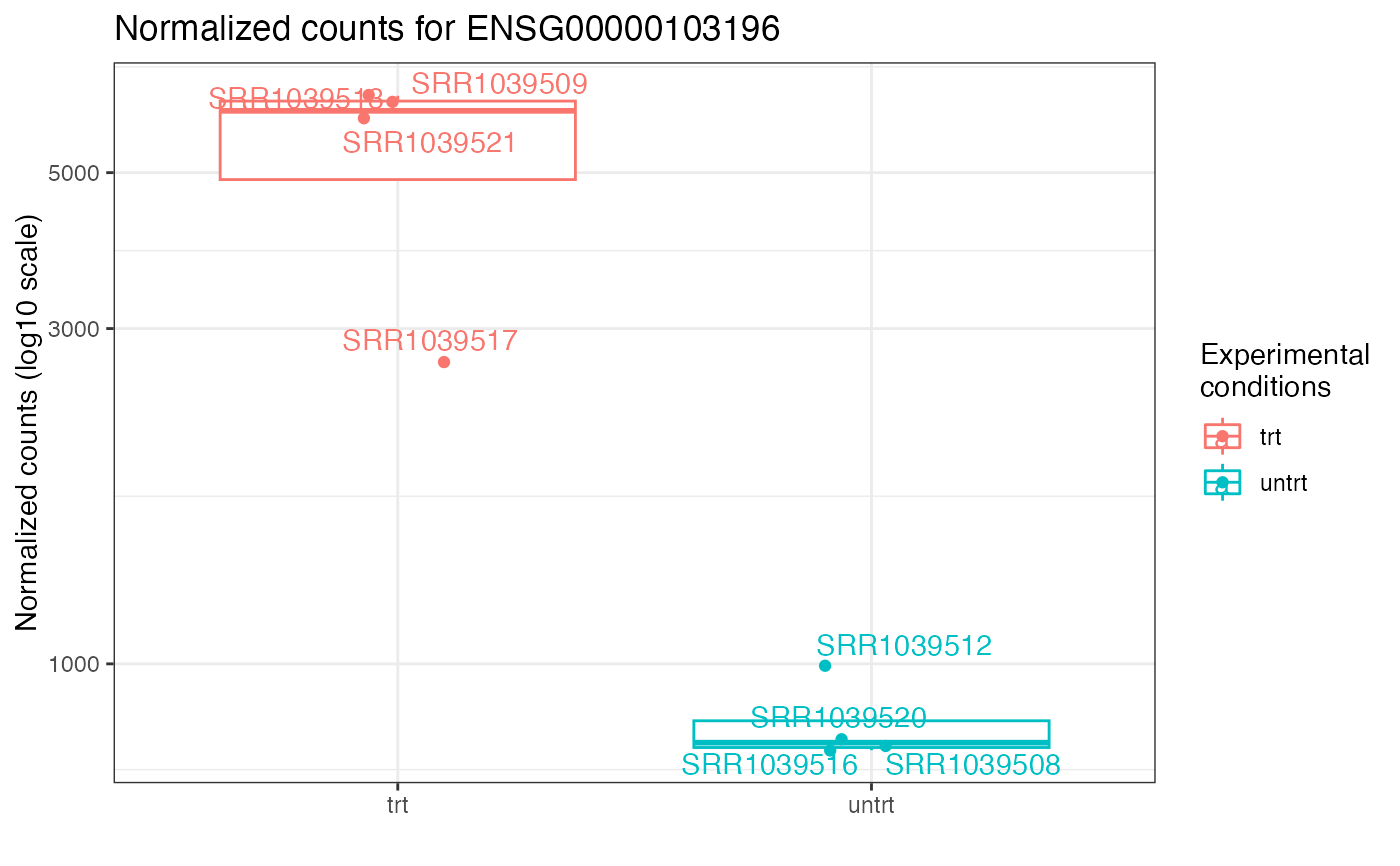
Plot for normalized counts of a single gene, with jittered points superimposed on the boxplot
ggplotCounts(
dds,
gene,
intgroup = "condition",
annotation_obj = NULL,
transform = TRUE,
labels_repel = TRUE
)Arguments
- dds
A
DESeq2::DESeqDataSet()object.- gene
A character, specifying the name of the gene to plot
- intgroup
Interesting groups: a character vector of names in
colData(dds)to use for grouping- annotation_obj
A
data.frameobject, withrow.namesas gene identifiers (e.g. ENSEMBL ids) and a column,gene_name, containing e.g. HGNC-based gene symbols. Optional.- transform
Logical value, corresponding whether to have log scale y-axis or not. Defaults to TRUE.
- labels_repel
Logical value. Whether to use
ggrepel's functions to place labels; defaults to TRUE.
Value
An object created by ggplot
Details
Note: this function relies on the DESeq2::plotCounts() function of DESeq2,
therefore pseudocounts of 0.5 are added to each point
Examples
library("airway")
data("airway", package = "airway")
airway
#> class: RangedSummarizedExperiment
#> dim: 63677 8
#> metadata(1): ''
#> assays(1): counts
#> rownames(63677): ENSG00000000003 ENSG00000000005 ... ENSG00000273492
#> ENSG00000273493
#> rowData names(10): gene_id gene_name ... seq_coord_system symbol
#> colnames(8): SRR1039508 SRR1039509 ... SRR1039520 SRR1039521
#> colData names(9): SampleName cell ... Sample BioSample
dds_airway <- DESeq2::DESeqDataSetFromMatrix(assay(airway),
colData = colData(airway),
design = ~ cell + dex
)
ggplotCounts(dds_airway,
gene = "ENSG00000103196", # CRISPLD2 in the original publication
intgroup = "dex"
)
#> Warning: Please use `mosdef::gene_plot()` in replacement of the `ggplotCounts()` function, originally located in the ideal package.
#> Check the manual page for `?mosdef::gene_plot()` to see the details on how to use it, e.g. refer to the new parameter definition and naming