Create an upset plot for genesets
gs_upset(
res_enrich,
res_de = NULL,
annotation_obj = NULL,
n_gs = 10,
gtl = NULL,
gs_ids = NULL,
add_de_direction = FALSE,
add_de_gsgenes = FALSE,
col_upDE = "#E41A1C",
col_downDE = "#377EB8",
upset_geom = geom_point(size = 2),
return_upsetgsg = FALSE
)Arguments
- res_enrich
A
data.frameobject, storing the result of the functional enrichment analysis. See more in the main function,GeneTonic(), to check the formatting requirements (a minimal set of columns should be present).- res_de
A
DESeqResultsobject.- annotation_obj
A
data.frameobject with the feature annotation information, with at least two columns,gene_idandgene_name.- n_gs
Integer value, corresponding to the maximal number of gene sets to be included
- gtl
A
GeneTonic-list object, containing in its slots the arguments specified above:dds,res_de,res_enrich, andannotation_obj- the names of the list must be specified following the content they are expecting- gs_ids
Character vector, containing a subset of
gs_idas they are available inres_enrich. Lists the gene sets to be included in addition to the top ones (vian_gs)- add_de_direction
Logical, whether to add an annotation with info on the DE direction of single genes
- add_de_gsgenes
Logical, if set to TRUE adds an annotation with detail on the single components of each defined subset
- col_upDE
Character, specifying the color value to be used to mark upregulated genes
- col_downDE
Character, specifying the color value to be used to mark downregulated genes
- upset_geom
A geom specification to be used in the upset chart. Defaults sensibly to
geom_point(size = 2)- return_upsetgsg
Logical, controlling the returned value. If set to TRUE, this function will not generate the plot but only create the corresponding data.frame, in case the user wants to proceed with a custom call to create an upset plot.
Value
A ggplot object (if plotting), or alternatively a data.frame
Examples
library("macrophage")
library("DESeq2")
library("org.Hs.eg.db")
library("AnnotationDbi")
# dds object
data("gse", package = "macrophage")
dds_macrophage <- DESeqDataSet(gse, design = ~ line + condition)
#> using counts and average transcript lengths from tximeta
rownames(dds_macrophage) <- substr(rownames(dds_macrophage), 1, 15)
dds_macrophage <- estimateSizeFactors(dds_macrophage)
#> using 'avgTxLength' from assays(dds), correcting for library size
# annotation object
anno_df <- data.frame(
gene_id = rownames(dds_macrophage),
gene_name = mapIds(org.Hs.eg.db,
keys = rownames(dds_macrophage),
column = "SYMBOL",
keytype = "ENSEMBL"
),
stringsAsFactors = FALSE,
row.names = rownames(dds_macrophage)
)
#> 'select()' returned 1:many mapping between keys and columns
# res object
data(res_de_macrophage, package = "GeneTonic")
res_de <- res_macrophage_IFNg_vs_naive
# res_enrich object
data(res_enrich_macrophage, package = "GeneTonic")
res_enrich <- shake_topGOtableResult(topgoDE_macrophage_IFNg_vs_naive)
#> Found 500 gene sets in `topGOtableResult` object.
#> Converting for usage in GeneTonic...
res_enrich <- get_aggrscores(res_enrich, res_de, anno_df)
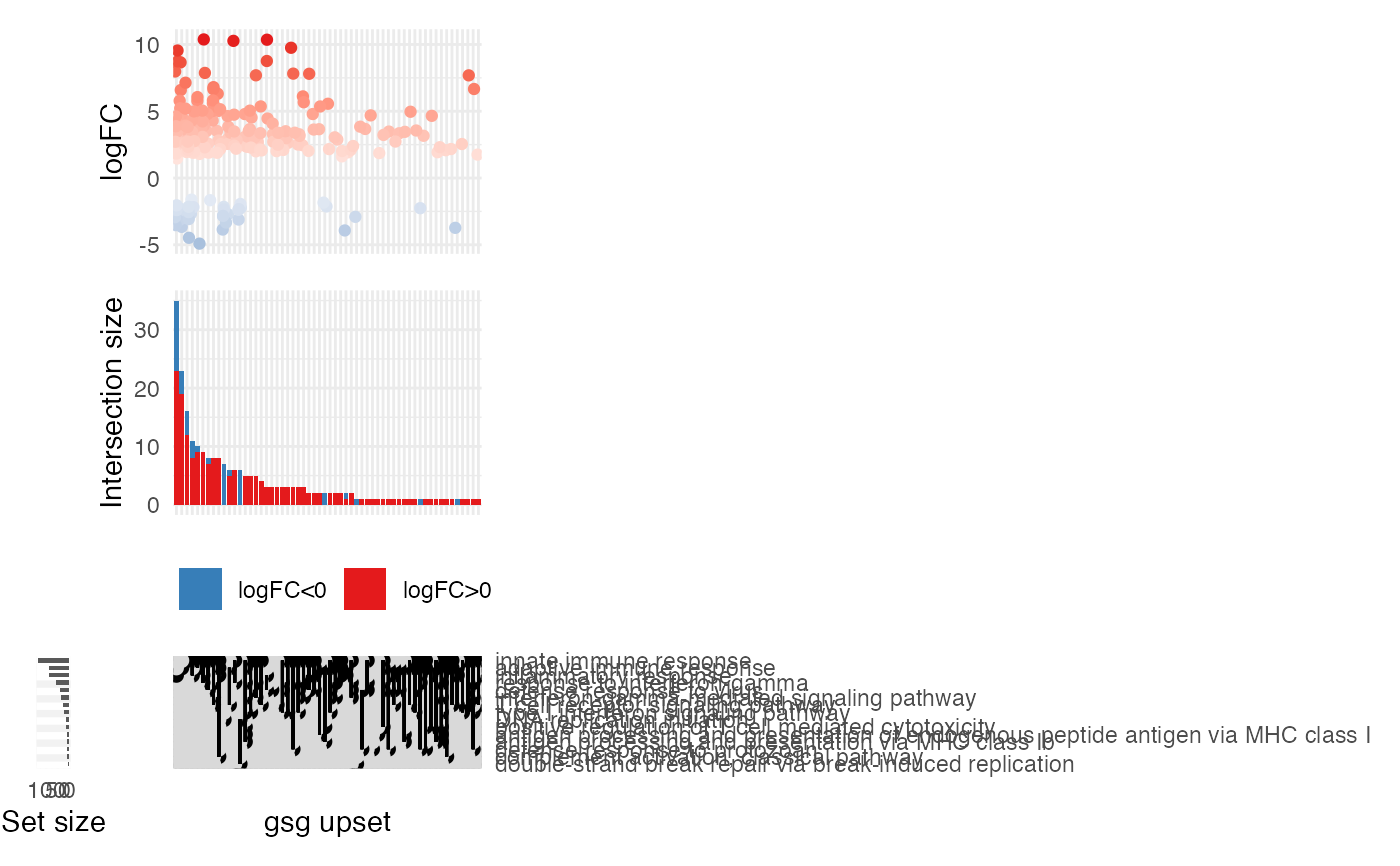
gs_upset(res_enrich,
n_gs = 10
)
#> Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
#> ℹ Please use `linewidth` instead.
#> ℹ The deprecated feature was likely used in the ComplexUpset package.
#> Please report the issue at
#> <https://github.com/krassowski/complex-upset/issues>.
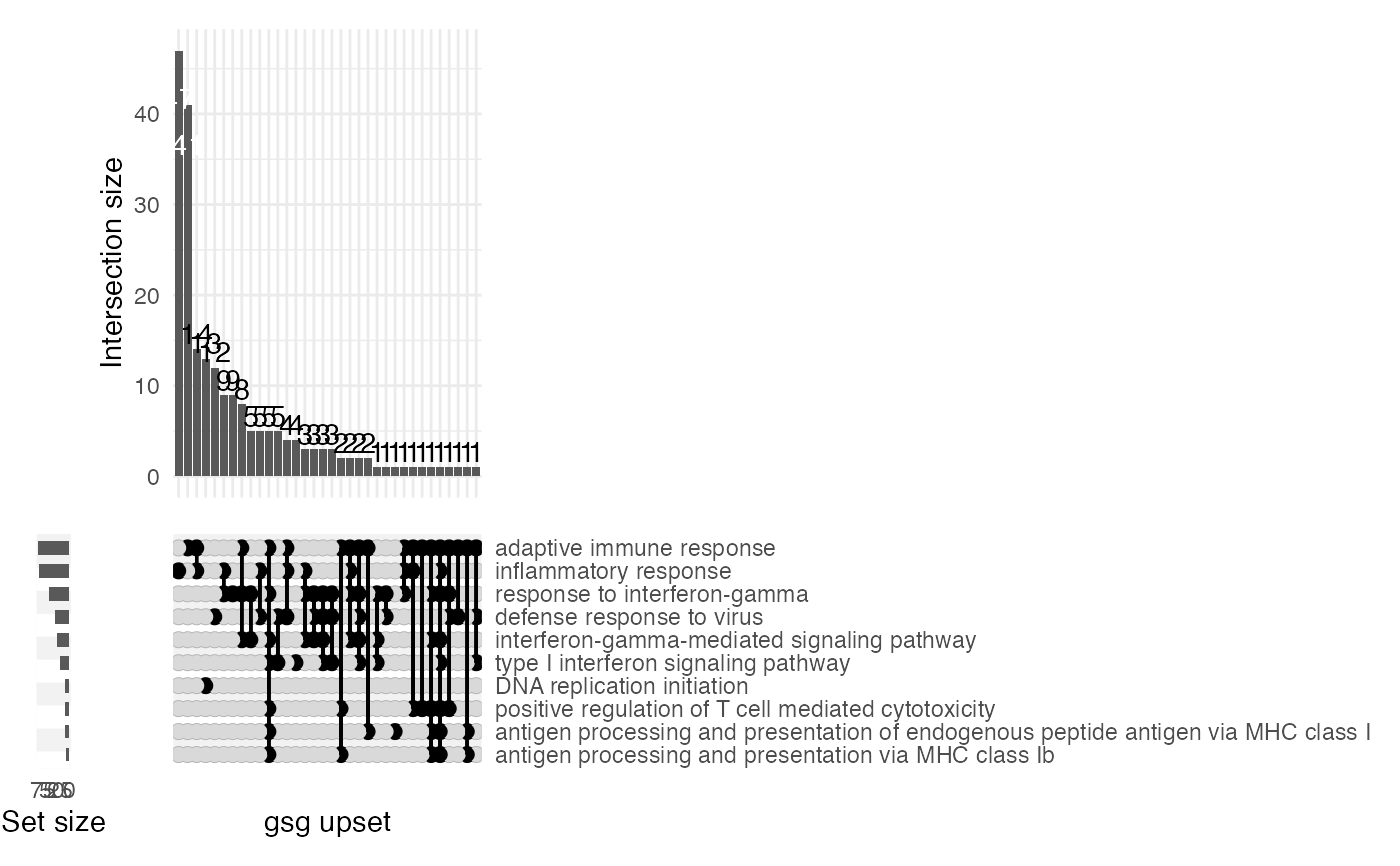 gs_upset(res_enrich,
res_de = res_de, annotation_obj = anno_df,
n_gs = 8,
add_de_direction = TRUE, add_de_gsgenes = TRUE
)
gs_upset(res_enrich,
res_de = res_de, annotation_obj = anno_df,
n_gs = 8,
add_de_direction = TRUE, add_de_gsgenes = TRUE
)
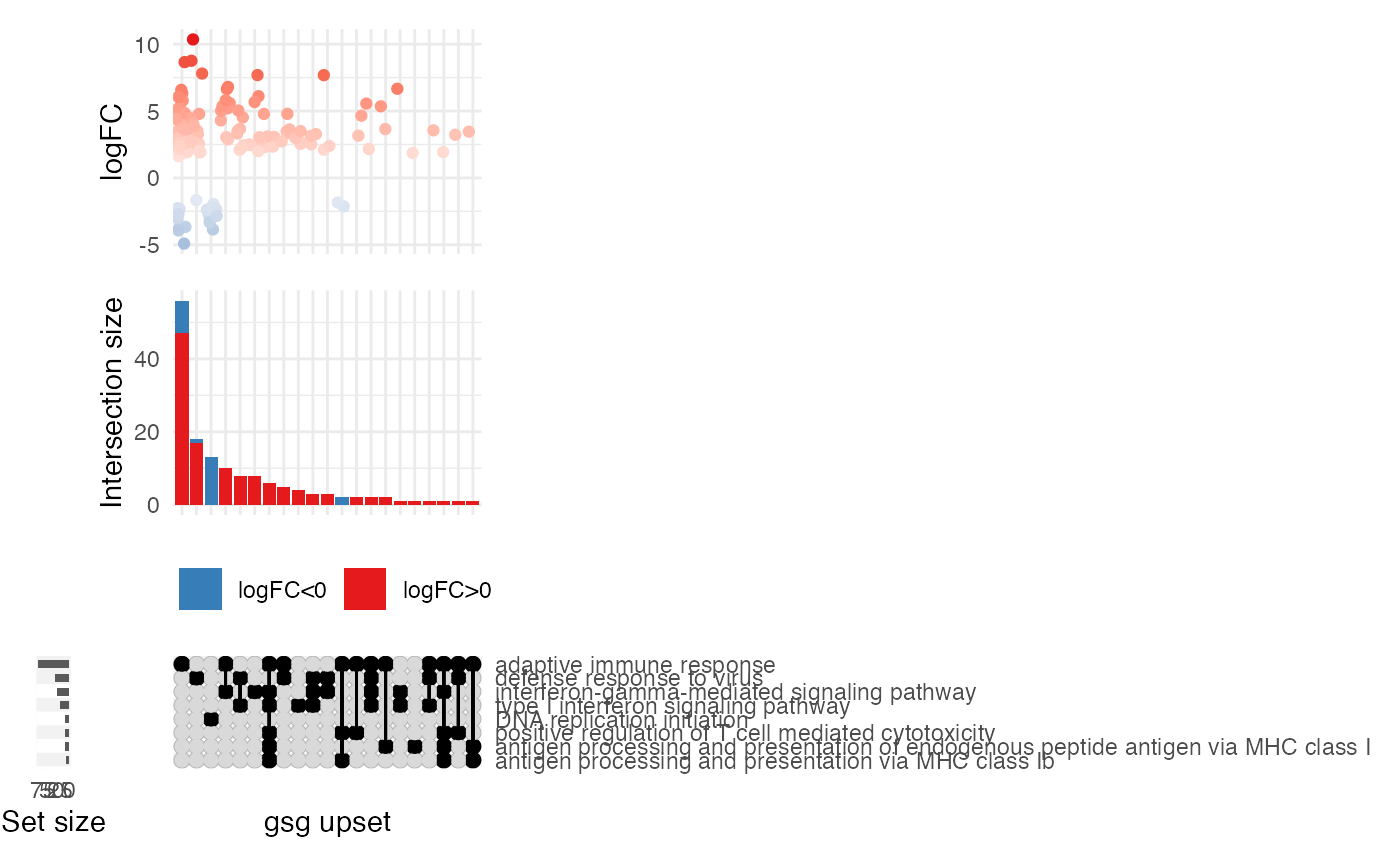 # or using the practical gtl (GeneTonicList)
gtl_macrophage <- GeneTonic_list(
dds = dds_macrophage,
res_de = res_de,
res_enrich = res_enrich,
annotation_obj = anno_df
)
#> ---------------------------------
#> ----- GeneTonicList object ------
#> ---------------------------------
#>
#> ----- dds object -----
#> Providing an expression object (as DESeqDataset) of 58294 features over 24 samples
#>
#> ----- res_de object -----
#> Providing a DE result object (as DESeqResults), 17806 features tested, 928 found as DE
#> Upregulated: 599
#> Downregulated: 329
#>
#> ----- res_enrich object -----
#> Providing an enrichment result object, 500 reported
#>
#> ----- annotation_obj object -----
#> Providing an annotation object of 58294 features with information on 2 identifier types
gs_upset(
gtl = gtl_macrophage,
n_gs = 15,
add_de_direction = TRUE, add_de_gsgenes = TRUE
)
# or using the practical gtl (GeneTonicList)
gtl_macrophage <- GeneTonic_list(
dds = dds_macrophage,
res_de = res_de,
res_enrich = res_enrich,
annotation_obj = anno_df
)
#> ---------------------------------
#> ----- GeneTonicList object ------
#> ---------------------------------
#>
#> ----- dds object -----
#> Providing an expression object (as DESeqDataset) of 58294 features over 24 samples
#>
#> ----- res_de object -----
#> Providing a DE result object (as DESeqResults), 17806 features tested, 928 found as DE
#> Upregulated: 599
#> Downregulated: 329
#>
#> ----- res_enrich object -----
#> Providing an enrichment result object, 500 reported
#>
#> ----- annotation_obj object -----
#> Providing an annotation object of 58294 features with information on 2 identifier types
gs_upset(
gtl = gtl_macrophage,
n_gs = 15,
add_de_direction = TRUE, add_de_gsgenes = TRUE
)